


中美歐臨床試驗用藥品生產質量管理規范對照
發布日期:2022-07-18 閱讀次數:11934 來源:中國醫藥報
摘要:

臨床試驗用藥品是指用于臨床試驗的試驗藥物、對照藥品、安慰劑,其生產質量管理是臨床試驗中的關鍵因素。
2022年5月,國家藥監局發布了《臨床試驗用藥品(試行)》附錄,自7月1日起正式施行。作為《藥品生產質量管理規范》(2010年修訂)的配套文件,該附錄旨在規范和指導臨床試驗用藥品制備,確保臨床試驗用藥品質量,保障受試者安全。
本文梳理分析了中國、美國、歐盟在臨床試驗用藥品生產質量管理方面的法規要求,供讀者參考。
□ 許丹
臨床試驗用藥品生產具有特殊性
臨床試驗用藥品一方面直接影響受試者的安全,另一方面其有效性決定了新藥臨床試驗結果;臨床試驗用藥品質量是否可控,也關系到新藥的評價和能否獲批上市。可以說,臨床試驗用藥品是新藥研發的關鍵產品,規范其生產質量管理非常重要,應最大限度降低研發生產環節引入的安全和質量風險。
不同于已上市藥品的生產,臨床試驗用藥品的生產具有特殊性。首先,臨床試驗用藥品生產處于藥物研發過程中,工藝和研究還不充分,生產批量小、批次少。而藥品生產質量管理規范(GMP)是針對藥品商業化生產規模的產品,其典型特征為商業化批量重復性生產,可能不完全適用于臨床試驗用藥品的生產。其次,在藥品早期研發階段,企業缺少對臨床試驗用藥品毒性的全面了解,在處方配比和生產工藝驗證上存在不完善的現象。再次,臨床試驗設計的多樣性對藥品生產、包裝與貼簽等方面提出更多的要求。比如生產用于雙盲試驗的臨床試驗用藥品,既需要包裝設計不能引起破盲,也要保證藥品的可追溯性,保證在緊急情況下揭盲的需要。此時不僅區別于商業化GMP的要求,更增加了出現混淆和差錯的風險。此外,臨床試驗用藥品處在產品研發階段,企業可能迫于對研發進度的追求采取快速且粗放(quick and dirty)的研發策略,而忽視生產質量管理。
臨床試驗用藥品生產的特殊性,決定了其生產管理不能完全照搬商業化藥品GMP管理模式,必須建立起一個高效的質量管理體系,兼顧臨床試驗用藥品的特殊性和不確定性,規范臨床試驗用藥品生產。因此,為指導企業建立臨床試驗用藥品生產質量管理體系,同時也為監管部門提供相關的監管依據,許多國家都制定并實施了臨床試驗用藥品生產質量管理規范(IMP),并在新藥注冊過程中按相應規范對臨床試驗用藥品生產質量管理情況進行現場檢查。
我國IMP法規要求
我國于2002年施行的《藥品注冊管理辦法(試行)》首次規定,“臨床研究用藥物,應當在符合《藥品生產質量管理規范》條件的車間制備。制備過程應當嚴格執行《藥品生產質量管理規范》的要求”;2007年施行的《藥品注冊管理辦法》保留了此項要求。新修訂《藥品注冊管理辦法》(2020年7月1日施行)規定,“藥物臨床試驗用藥品的管理應當符合藥物臨床試驗質量管理規范的有關要求”,同期實施的《藥物臨床試驗質量管理規范》規定了“試驗藥物的制備應當符合臨床試驗用藥品生產質量管理相關要求”。因此,一部“臨床試驗用藥品生產質量管理規范”亟待制定。
在藥品全生命周期監管中,產品研發、臨床試驗、商業化生產、流通和退市等每一環節應當有對應的法規要求,從而統一行業行動標準,提升整體水平。目前我國藥品監管法規體系中,藥品商業化生產應遵守《藥品生產質量管理規范》,藥品研發階段的臨床使用環節應遵守《藥物臨床試驗質量管理規范》。為規范和指導臨床試驗用藥品制備,支持研究和創制新藥,參考相關國際做法,國家藥監局于2022年5月發布了《臨床試驗用藥品(試行)》,以避免在臨床試驗用藥品制備過程中由于不良生產行為引入的安全和質量問題,最大限度降低生產環節引入風險,保障臨床試驗用藥品質量安全。
美國IMP法規要求
美國食品藥品管理局(FDA)最早在1997年發布了《新藥臨床試驗用樣品制備技術指導原則》,其中規定了臨床試驗用藥品生產時必須符合cGMP的規范。該文件也談到了臨床試驗用藥品的生產和商業化生產的不同,為了保證受試者的安全,臨床試驗用藥品需要經常接受FDA的cGMP現場檢查。
為了區分臨床試驗用藥品對cGMP要求的符合程度不同,FDA于2008年7月發布了《Ⅰ期臨床試驗用藥品生產質量管理指導原則》,全文強調了Ⅰ期臨床試驗所用的研究藥品生產必須符合cGMP的要求,同時為了兼顧研究藥品的特殊性,部分生產管理可以參照該指導原則執行,給臨床試驗用藥品生產帶來了一些靈活性;同時也說明了生產廠家可以采用其他代替方法達到該指南的要求。該指導原則主要包括人員、質量控制、機構與設備、藥品組分和包裝的控制、生產和記錄、實驗室控制、包裝貼標簽和發送、記錄保存等方面。
與歐盟2010年發布的歐盟GMP附錄13《臨床試驗用藥品》不同,FDA發布的指導原則沒有突出臨床試驗用藥品設盲需求對生產管理帶來的影響。對于歐盟重點強調的標簽部分,FDA在該指導原則中相對涉及較少。
FDA在2003年發布了《Ⅱ期和Ⅲ期臨床試驗用藥品的指導原則》,其中更多地強調Ⅱ期和Ⅲ期注冊申報資料的要求,對臨床試驗用藥品的生產和質量控制的管理涉及較少。
歐盟IMP法規要求
歐盟在臨床試驗指令(Directive 2001/20/EC)中對臨床試驗用藥品進行了定義,規定其為用于臨床試驗的具有活性物質的藥物或安慰劑形式的藥物,同時也包括了已經上市的陽性對照藥品。
2014年,歐盟發布了人用藥臨床試驗法規(Regulation(EU)No 536/2014),其中第九章“臨床試驗用藥品和輔助藥品的生產和進口”中,單獨強調了臨床試驗用藥品的通用要求。如第62條指出,需要指定符合相應資質的質量負責人(Quality Person,QP),負責對每一批臨床試驗用藥品進行放行。但是對其與商業化生產和質量控制異同沒有詳細規定。同時,第十章“標簽”中明確了臨床試驗用藥品的標簽中應當有如下內容:臨床試驗負責人信息、臨床試驗信息,以及藥品相關信息。
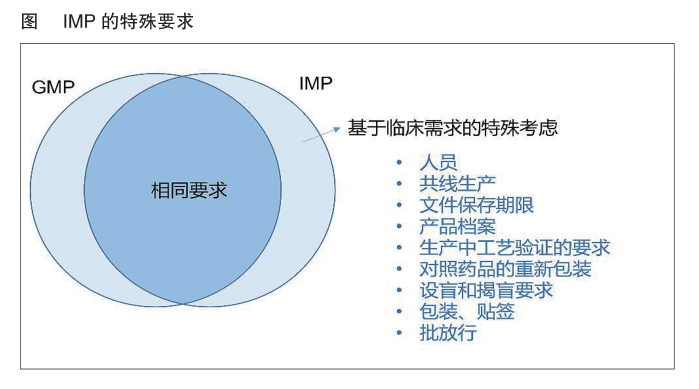
2010年發布的歐盟GMP附錄13《臨床試驗用藥品》,專門對臨床試驗用藥品的生產質量管理提出了具體要求,較上述法令和法規更為詳細。該附錄共分15個章節,包括原則、質量管理、人員、廠房與設備、文件管理、生產、盲法操作、隨機編碼、包裝、貼標、質量控制、批放行、運輸、投訴、召回與退貨。
附錄13總體框架和歐盟GMP正文類似,但是除去與商業化生產相同的要求,結合臨床試驗用藥品的特殊性(設盲和可追溯性),重點強調了廠房設施、生產管理、盲法操作、包裝和貼標等的不同點。如在公用廠房和設施部分,由于對臨床試驗用藥品的毒性、效力與潛在敏感性可能未全面了解,所以更需要使交叉污染風險最小化,推薦盡可能采用專線生產方式;如果沒有專線,推薦采用階段化生產方式。在生產控制方面,由于早期工藝尚未充分,可以放寬對產品工藝驗證的要求;但是對無菌和病毒滅活/ 去除工藝需要進行證明,確保臨床試驗用藥品質量安全。在設盲操作時,既要保證符合GMP的可追溯性,同時也要滿足盲法試驗不破盲的要求,因此該附錄對標簽的管理方式也提出了詳細要求。
同時,歐盟《原料藥生產質量管理規范》中的第19章“用于臨床研究的原料藥”部分,也詳細規定了用于生產臨床試驗用藥品的原料藥的管理要求。該章節強調了在臨床試驗用藥品生產早期,原輔料質量標準尚未確定,在保障受試者安全的前體下,可以放寬對原輔料質量控制的要求,原料藥可以只憑供應商的檢驗報告即可采用。與工業化生產相比,臨床試驗用藥品的原料藥產量不確定性高、波動性大,因此通常不要求對產量的變化進行調查。由于臨床試驗用藥品的原料藥工藝為固定僅生產一批,或開發過程需不斷進行工藝變更,因此對于原料藥的工藝驗證也沒有商業化生產的要求。生產過程中的偏差,不需要調查到根本原因,只需要如實記錄。
中美歐IMP法規對比分析
總體來說,我國和歐盟的IMP要求更相似,但鑒于我國的實際情況有一些特殊考慮。例如在文件體例上,由于我國《臨床試驗用藥品(試行)》的定位為《藥品生產質量管理規范》的第13個附錄,因此對于原料藥的特殊要求融入到正文“物料”章節中,不再以類似歐盟IMP的寫作方式,將原料藥另以附錄形式來另成一篇進行要求。在根據臨床試驗階段進行分期要求的問題上,我國《臨床試驗用藥品(試行)》和歐盟要求一致,淡化臨床試驗分期的區別,即無論是臨床試驗早期還是確證性臨床試驗階段,臨床試驗用藥品生產均應最大程度上符合GMP的通用要求,不再有區別對待;但是考慮在臨床試驗早期一些初創企業不具備完備GMP條件,允許其采取委托生產的方式進行臨床試驗用藥品的生產。此外還有其他具體差異,詳見表格。
對于實施IMP后藥監部門是否會針對臨床試驗用藥品生產管理進行核查的問題,歐盟在2001/20/EC指令第15(1)款中提到,可以由被藥監局認可的第三方機構進行核查;而FDA更傾向于在生產場地進行生產許可檢查同時進行臨床試驗用藥品檢查。 我國《臨床試驗用藥品(試行)》中未涉及檢查的內容,但是在《藥品注冊核查要點與判定原則(藥學研制和生產現場)(試行)》(2022年1月1日施行)中規定研制現場核查“必要時,可前溯至研究立項、處方篩選、工藝優化等研究內容”。
綜上,我國新發布的《臨床試驗用藥品(試行)》填補了監管法規體系的空白,其在適用范圍和管理要求等方面與歐美法規有很多相似之處,同時結合我國國情也存在一定特殊考慮。該附錄的正式施行,將推動臨床試驗用藥品生產規范化,同時兼顧了臨床試驗用藥品的特殊需求,規范臨床試驗用藥品生產質量管理,保護臨床受試者的權益,保障臨床試驗的質量。
相關閱讀
WHO的IMP法規要求
1996年,世界衛生組織(WHO)發布了Technical Report Series 863附錄7《人用臨床試驗用藥品生產質量管理規范》,該附錄強調了臨床試驗用藥品生產的通用要求,包括質量保證、驗證、投訴、召回、人員、廠房和設施、物料、文件、生產管理、質量控制、發運、退回和銷毀等方面。其表述內容和歐盟IMP的要求基本相同,突出強調了文件管理的內容,具體包括生產訂單、產品檔案、標準、工藝規程、包裝、標簽、重新貼標簽、設盲編碼等要求。
由于WHO對COVID-19治療藥物進行檢查需要新的指導原則,WHO第55次藥物制劑專家委員會(ECSPP)同意了預認證-檢查小組(PQT INS)提出的緊急修訂臨床試驗用藥品GMP指南的提案。2020年底,WHO發布臨床試驗用藥品生產質量管理規范草案,該草案是對1996年版IMP的更新,于2021年1月開始公開征求意見。該草案共18個板塊,涵蓋了藥品生產質量管理全過程,涉及的具體領域包括質量管理、確認和驗證、外包活動、自檢和質量審核以及人員培訓。同時,場地、設備、儀器和材料、文件記錄、加工和工藝設計、質量控制、穩定性等相關考慮因素也包含在指南草案中。修訂該指南的主要目的,是為了其與當前國際上對GMP的期望和趨勢保持一致,并與其他國際指南中的原則保持一致。因此,具體內容和歐盟要求保持一致,但是并未和FDA一樣對臨床試驗用藥品有Ⅰ期、Ⅱ期和Ⅲ期分期的要求。
需要注意的是,WHO作為一個國際協調機構,還負責對各個國家的藥品監管機構進行評估和指導,其中WHO的NRA(National Regulate Assessment)評估項目中CT01.04指標要求“臨床試驗用產品符合臨床試驗用產品生產質量管理規范的法律條款、法規和指南”。而在2014年WHO對我國的評估中,我國藥品監管法規中缺少該部分內容。因此,我國新發布的《臨床試驗用藥品(試行)》,填補了該項法規空白,也標志著我國藥品監管更加成熟完善。
相關新聞

醫學科普,聽得懂更要講得對
新一輪科技革命推動醫學科技迅速發展,新裝備、新技術、新藥、新方案等已深度影響“促、防、診、控、治、康”各環節,這也為健康科普提供了高水平的傳播內容和傳播載體。醫學科普是以通俗易懂的方式將健康領域的科技知識、科學方法、科學思想和科學精神傳播給公眾,旨在培養公眾的健康素養,幫助公眾學會自我健康管理的長期性活動。建設健康中國,醫學科普工作具有重要意義和獨特作用。
9794個小時之前
基于質量源于設計路線的生物類似藥質量研究
24842個小時之前
淺談AI技術在COVID-19診療中的應用
25009個小時之前
“OK鏡”市場迎來變數?療效及安全備受關注
25009個小時之前









